

Single Nucleotide Variants Analysis
Single nucleotide variants (SNVs) occur when a single nucleotide in the DNA sequence (e.g., A, T, C, or G) is altered. SNVs are the most common type of sequence change in the human genome and play an important role in disease susceptibility and an individual's response to therapy. There are many sources of endogenous and exogenous damage that result in single base pair substitution mutations that produce SNVs. Like SNVs, single nucleotide polymorphisms (SNPs) are single base substitutions, but they are restricted to germline DNA and must be present in at least 1% of the population.
A large number of SNVs have been identified through genome-wide association studies (GWAS) in association with a variety of human diseases and disorders, and the biological impact of SNVs in coding regions depends on their type (synonymous vs. missense), and in non-coding regions on their impact on RNA processing or gene regulation. These disease-specific discoveries are expected to facilitate the development of precision medicine. However, the intrinsic effect of single nucleotide changes on the overall physicochemical properties of the nucleic acid sequence is minimal, making SNV detection a challenging task.
Next-generation sequencing (NGS) technologies have a groundbreaking ability to detect SNVs with little or no prior knowledge of the location or identity of the variant in the DNA sequence. However, in these technologies, the vast majority of sequencing data maps to non-informative wild-type sequences, which increases the time and cost of analysis. In addition, the large volume of data generated by next-generation sequencing and the complexity of its analysis are incompatible with routine clinical applications Widespread implementation of precision medicine requires simpler, faster, and more cost-effective technologies capable of targeted detection of SNVs at the point of care (POC). In recent years, long-read sequencing technologies, including Pacific Biosciences and Oxford Nanopore, have provided an opportunity to address these challenges by enabling the detection and phasing of small variants at the single-molecule level without the need for amplification.
Major Approaches for SNV Detection: from Array to the Latest Sequencing Technologies
Microarray Technology
Microarrays have been a mainstay of the genomics field for many years, providing a proven method for analyzing SNVs. They are particularly well suited to detecting known SNPs in specific genomic regions. However, while microarrays offer a powerful data analysis process, they are limited in that they can only detect known SNPs. Novel SNVs, which are increasingly important for groundbreaking genetic research, may escape their notice.
Advantages:
- Data quality and track record: Time-tested and trusted microarrays provide consistent and reliable results.
- Scalability: Microarrays are particularly effective for large population studies, allowing efficient detection of known SNVs in large numbers of samples.
- Target-specific: The technology can be focused on specific genomic regions to pinpoint relevant SNPs.
Next-generation Sequencing (NGS)
NGS provides a broader, more detailed view of the genome. The utility of clinical NGS assays designed to detect SNVs is dependent on assay design features, including amplification-based versus hybrid capture-based targeting methods, DNA library complexity, sequencing depth, tumor cellular architecture (in cancer sample sequencing), specimen fixation, and sequencing platform. These traditional sequencing technologies typically require the use of polymerase chain reaction (PCR), which limits SNV detection to regions that can be easily amplified. In addition, while these methods can detect a large number of variants, their limited read lengths often limit the quantification of these variants.
Advantages:
- Unbiased Variant Discovery: NGS is not constrained by a priori knowledge about SNV, making it well suited for discovering unknown variants.
- High throughput and scalability: Its ability to sequence a large number of samples simultaneously is unrivaled.
- Small sample size: NGS requires minimal DNA input to retain valuable samples.
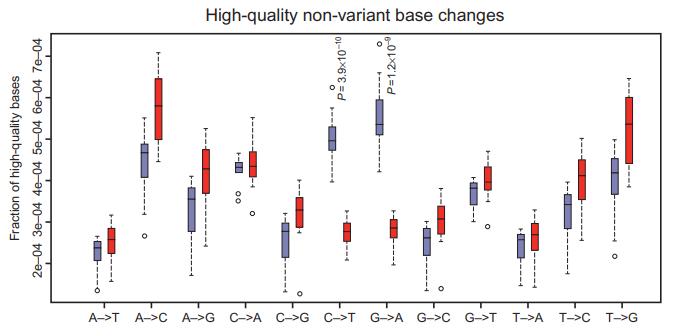
Observed spectrum of high-quality base changes in FFPE and frozen NGS data. (Spencer et al., 2015)
Long-read Sequencing Technology
Long-read sequencing technologies, such as those offered by Pacific Biosciences and Oxford Nanopore, provide a viable solution to the limitations of short-read long sequencing. These technologies have the potential to sequence more than 100 billion bases in a single run and generate reads in excess of 10kb in length, providing unparalleled depth and resolution.
Advantages:
- Enhanced phasing: The technology provides extended sequencing reads with unparalleled background, which is critical for phasing and interpreting genetic data.
- Eliminates PCR: The innate ability to sequence natural DNA eliminates the need for the polymerase chain reaction (PCR), which has traditionally been an important step in sequencing DNA replicates.
- Real-time analysis: The ability to provide immediate results makes it invaluable when time is of the essence.
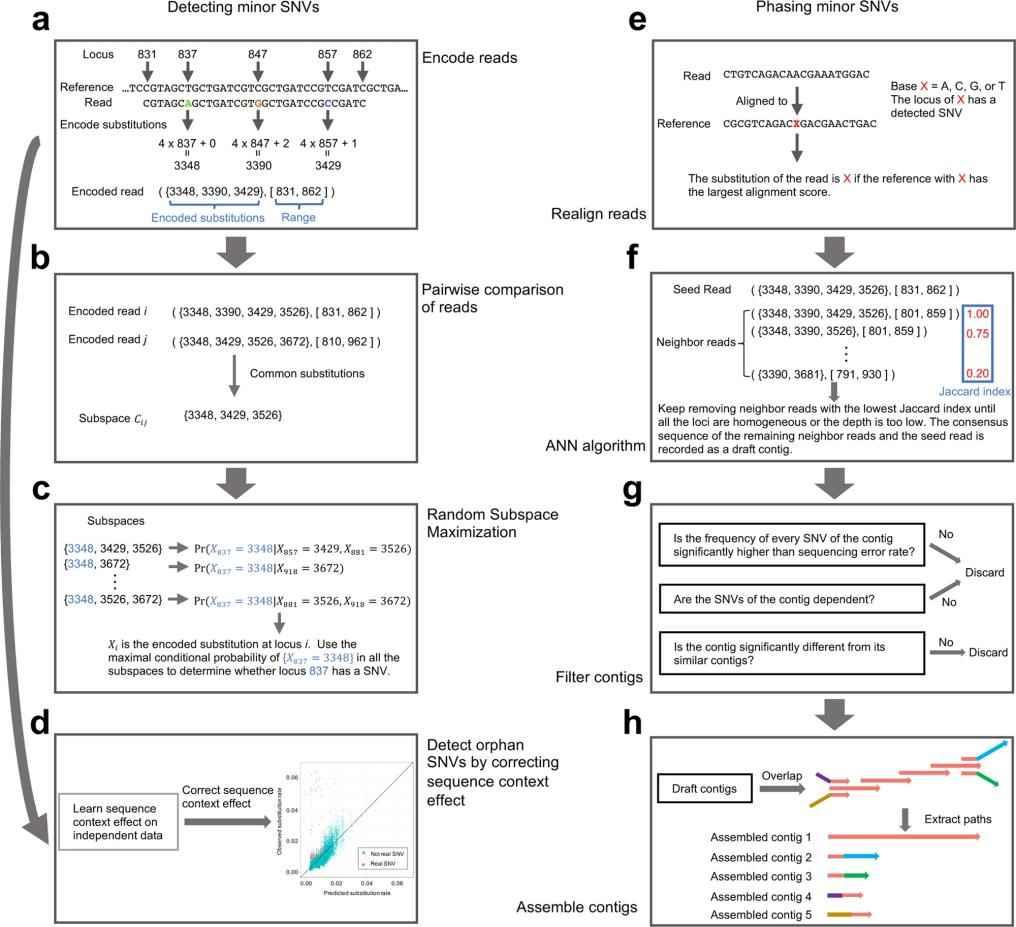
GDA (In Vivo Genomic Diversity Analyzer): A New Software Tool for SNV Analysis
With the need for a more accurate and comprehensive tool to detect and characterize minor SNVs, iGDA positions itself as an innovative solution in the field of genomics. Designed to operate on raw long-read sequencing data, iGDA can detect SNVs at frequencies as low as 0.2%.
One of iGDA's distinguishing features is its use of the Random Subspace Maximization (RSM) algorithm, which allows it to leverage information from multiple loci without being hindered by the combinatorial explosion. In addition, its adaptive nearest neighbor clustering (ANN) algorithm facilitates SNV phasing without the need to assume a fixed number of haplotypes.
Extensive testing on pooled long-read long sequencing datasets confirms the strength of iGDA. The tool detects 85.8% to 96.7% of true SNVs with a false discovery rate of less than 1%. In addition, its ability to stage minor SNVs showed an average accuracy range of 90.7% to 98.7%.
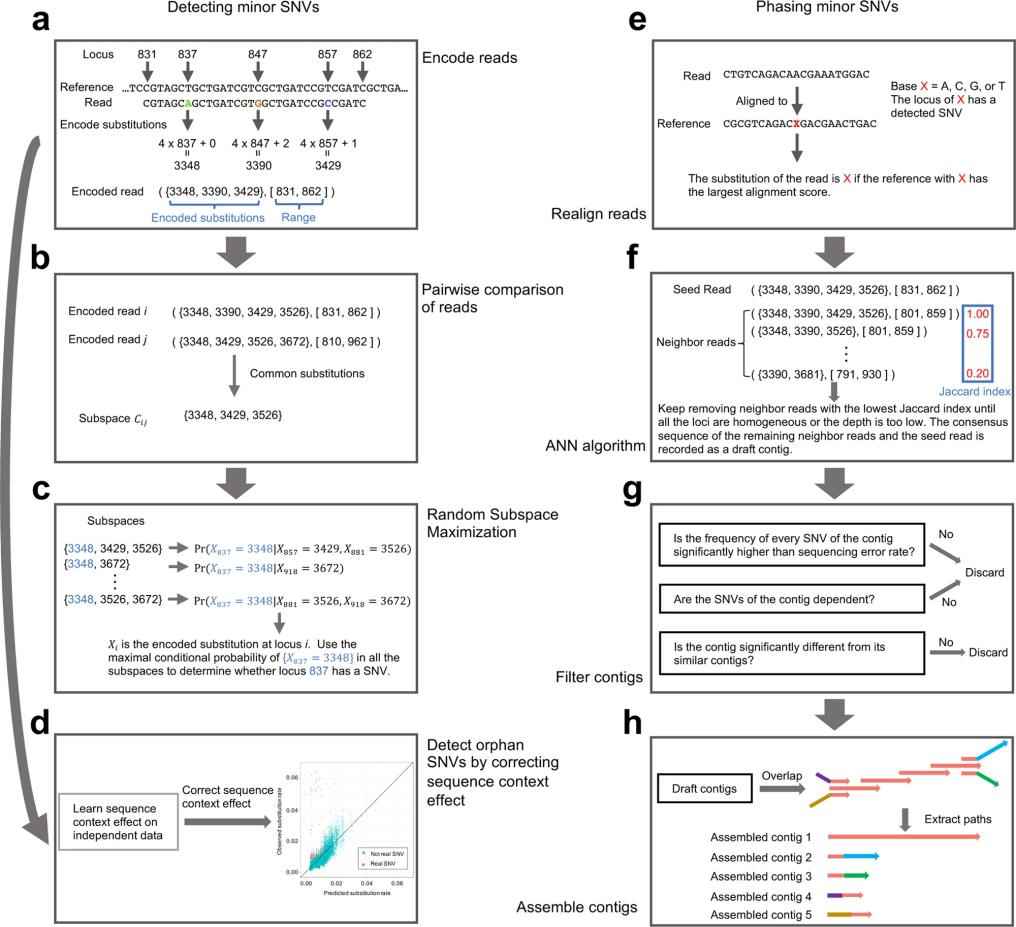
The main steps of iGDA. (Feng et al., 2021)
References
- Feng, Zhixing, et al. "Detecting and phasing minor single-nucleotide variants from long-read sequencing data." Nature communications 12.1 (2021): 3032.
- Spencer, David H., Bin Zhang, and John Pfeifer. "Single nucleotide variant detection using next generation sequencing." Clinical genomics. Academic Press, 2015. 109-127.