

Overview
The advent of long-read sequencing technologies has led to tremendous advances in crop genomics, and they help researchers provide comprehensive sequence data by detecting high-quality reference genomes. These advances provide reliable gene editing design options for crop improvement and can be used to validate constructs, confirm edits, and assess off-target effects.
Next-generation sequencing and short-read sequencing technologies have enabled a deeper understanding of plant species diversity, primarily at the single nucleotide polymorphism (SNP) level, but representing only a small fraction of the species-wide genomic space. A wide range of large chromosome-scale structural variants (SVs) ranging from hundreds of kilobases all the way up to a few megabases have been found to be associated with key agronomic traits in plant genomes. SVs have a strong impact on trait variation in plant species and may have a higher functional significance than SNPs. Due to the complexity and highly repetitive nature of plant genomes, this makes SV difficult to assess using high-throughput genomic screening methods. The development of long-read sequencing technologies (Pacific Biosciences and Oxford Nanopore sequencing) has transformed crop genomics from the era of a single reference genome to one in which we now have access to tens or hundreds of reference-quality genome assemblies within a species. Studies of SV and pan-genomic characterization within species can reveal extensive genome content variation among individuals within species, a paradigm shift in crop genomics and improvement.
Importance of Structural Variants in Crop Improvement
There are many mechanisms by which structural variation can be generated. For example, transposable elements (TEs) can self-replicate in the genome or they can capture gene sequences and carry them to new genomic locations. This process can lead to significant disruption of coding portions of the genome. In addition, errors during meiotic recombination may introduce structural variants, such as non-allelic homologous recombination and double-strand break repair by single-strand annealing. Finally, presence-absence variants (PAVs) can be created by differential genome segregation across genotypes following a genome-wide replication event, especially in plants.
Plant genomes, including crop species, are particularly enriched in TE, and the relevance of TE to crop phenotypes has been repeatedly demonstrated.TE can be functionally correlated in a variety of ways, including modification of the structure and number of transcribed gene products. As pangenomes are widely used in crop species, TE will receive increasing attention as a driver of structural variation in crop improvement.
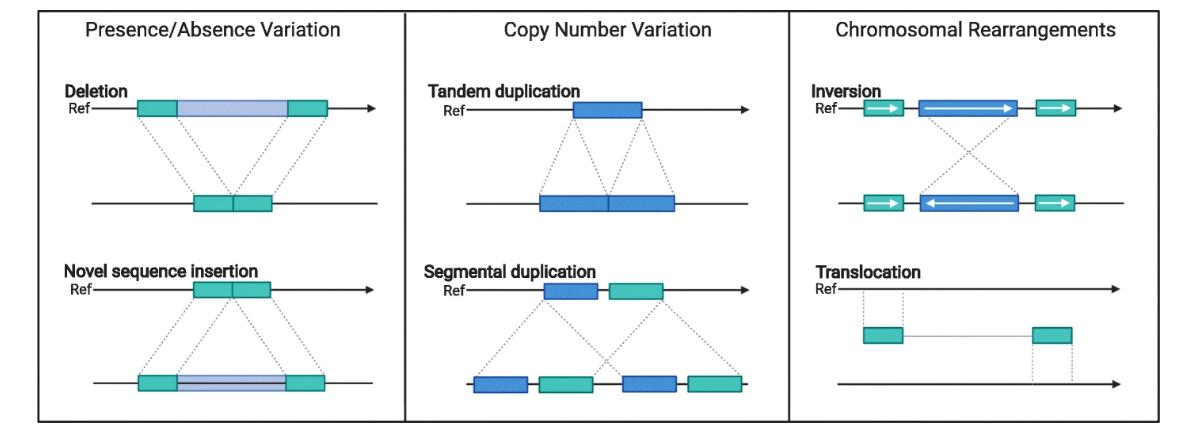
Diagrams of structural variants that can be found in crop genomes. (Della Coletta et al., 2021)
Long-read Sequencing for Crop Pan-Genomics Characterization and Improvement
Uncovering hidden genetic treasures
Traditional approaches that rely on a single reference genome limit our ability to capture the genome-wide diversity that exists within a species. In contrast, the concept of pan-genomes encapsulates the entirety of genetic information within a species, revealing richer dimensions of crop genomes. Long-read sequencing technologies such as PacBio and Oxford Nanopore provide the resolution needed to construct high-quality pan-genomes. For example, a wheat pangenome constructed using long-read sequencing revealed approximately 30,000 new genes not present in the reference genome, emphasizing the enormous genomic variation hidden in many crops.
Addressing polyploidy and structural variation
Polyploid genomes are prevalent in many crops and pose significant challenges due to their complexity and size. Long-read sequencing is able to span large repetitive regions, ensuring accurate phasing and identification of homologous sequences. For example, high-quality assembly of the peanut genome from the tetraploid crop became feasible thanks to long-read sequencing, revealing unique subgenomes and their interactions.
TEs have traditionally been considered as genomic "junk" but have recently gained recognition for their role in shaping gene function and regulating expression. Through long-read sequencing, we can accurately localize and annotate TEs, providing insights into their evolutionary impact. For example, it has been found that certain TEs in maize can influence flowering time, a trait of great agronomic importance. Beyond mere gene regulation, TEs often act as hotspots for generating genomic novelty. TE activity may lead to gene duplication, disruption, or new modes of regulation. In crops such as rice, TEs have been associated with key traits such as drought tolerance, grain size, and disease resistance, showing their potential for crop improvement.
Advancing quantitative trait locus (QTL) mapping and genome-wide association studies (GWAS)
QTL mapping of biparental populations and GWAS of different groups of individuals are used to identify genomic regions associated with desired phenotypes. Traditional QTL mapping and GWAS are limited by the use of a single reference genome. A pan-genomic approach supported by long-read sequencing allows researchers to utilize the comprehensive genetic pool of a species. This approach has been demonstrated in soybean, where a genome-wide GWAS revealed novel loci associated with oil content that were previously masked in single-reference studies.
Advancing genomic prediction and improvement
Many important traits for crop improvement are controlled by a number of low impact QTL. Complex genetic structure makes it difficult to identify all QTL behind a trait, correctly estimate their impacts, and use methods such as marker-assisted selection to infiltrate them into superior lines . Genomic selection is an alternative approach for complex traits in which marker effects are estimated from a training set, individual phenotypes are predicted based on the estimated marker effects (i.e., genomic prediction), and selection is based on the predicted phenotypes. Using SNPs as predictors, important agronomic traits such as grain yield, grain moisture, grain quality, biomass traits, and stem and root collapse can be predicted with fairly high accuracy. However, using a single reference for such applications introduces many limitations and biases.
Novel methods for identifying markers within a pan-genomic framework are needed to improve prediction accuracy. Practical haplotype mapping (PHG) is one such method that successfully handles the pan-genomic complexity of species at the scale required for complex traits and plant breeding programs]. In the PHG approach, the existing genomic resources of the founding lines of the breeding program are loaded into the graph pan-genomic database. Accurate estimation of individuals with low sequence coverage in breeding populations is achieved based on consensus haplotypes derived from the graph pan-genome database.PHG is a promising strategy to reduce genotyping costs while capturing broader diversity in large breeding populations.
Advancing gene editing
The detailed structural insights provided by pan-genomes can serve as a roadmap for precision genome editing efforts. Tools such as CRISPR/Cas9 can be used more effectively when full-spectrum genomic variation is understood. This precision allows for targeted crop improvement, whether it's improving nutrient content, enhancing disease resistance, or optimizing growth traits.
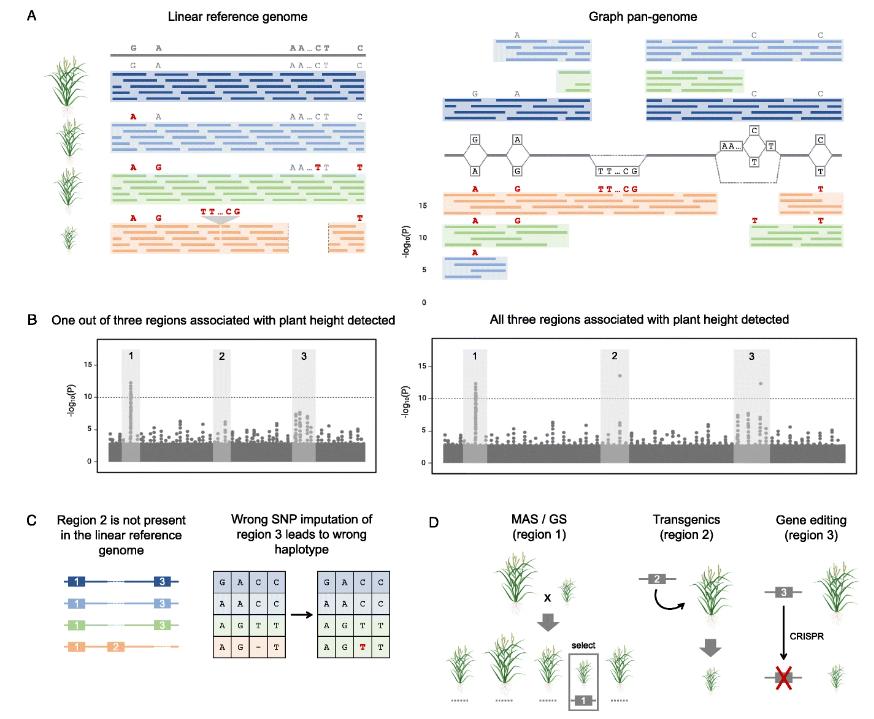
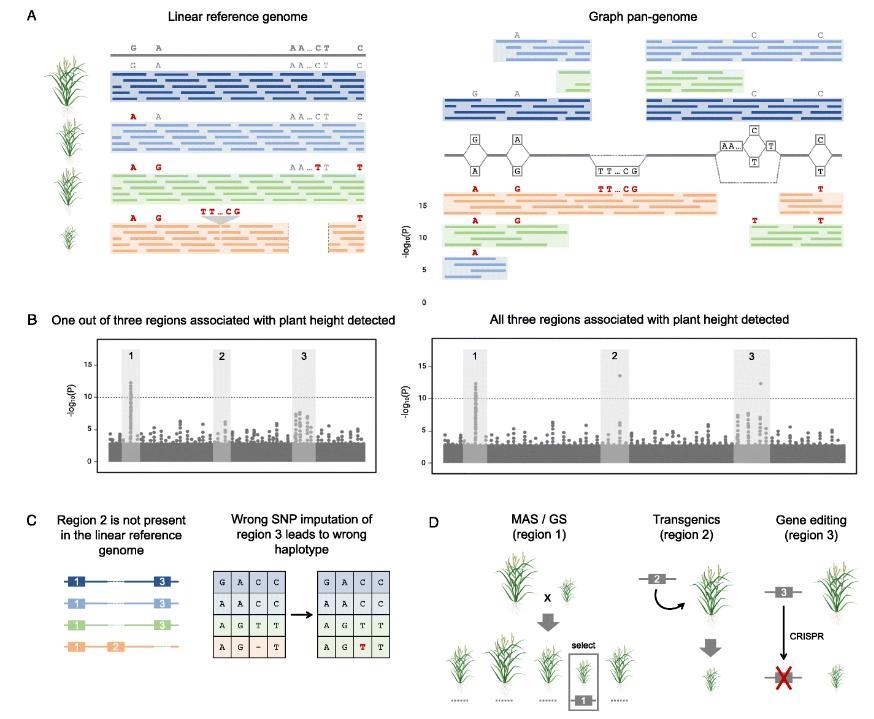
Impact of pan-genome representation on dissection of quantitative variation and applications to crop improvement. (Della Coletta et al., 2021)
Examples of Long-read Sequencing for Crop Genomics and Improvement
Kale-type oilseed rape, often referred to as canola, proves the strength of long-read, long-sequencing. This heteropolyploid crop has an inherently complex genome that poses significant challenges for genomics. However, when long-read sequencing was performed, a surprising revelation was made as much as 10% of all genes were affected by small to medium-sized structural variants. Even more surprisingly, nearly half of these variants ranged from 100 bp to 1,000 bp, a range that is often elusive to short-read sequencing methods such as Illumina.
Cassava, a staple food in tropical and subtropical regions, exemplifies the success of combining long-read sequencing with genome engineering. Conventional breeding techniques are cumbersome because cassava is resistant to genetic transformation. However, the use of CRISPR-Cas9 and long-read sequencing allows scientists to bypass these challenges and introduce targeted genetic modifications. These modifications are not arbitrary. They aim to give cassava valuable properties observed in other crops. One such effort was to infuse cassava with starch properties that do not contain straight-chain amylose, which has desirable cooking and processing properties. The combination of targeted mutagenesis and long-read sequencing has led to the development of cassava lines with varying levels of straight-chain amylose content. More importantly, these improved cassava lines retained their genetic integrity, making them GM-free.
References
- Della Coletta, Rafael, et al. "How the pan-genome is changing crop genomics and improvement." Genome biology 22.1 (2021): 1-19.
- Chawla, Harmeet Singh, et al. "Long‐read sequencing reveals widespread intragenic structural variants in a recent allopolyploid crop plant." Plant biotechnology journal 19.2 (2021): 240-250.
- Qi, Weihong, et al. "The haplotype-resolved chromosome pairs of a heterozygous diploid African cassava cultivar reveal novel pan-genome and allele-specific transcriptome features." GigaScience 11 (2022): giac028.