

Within the domain of high-throughput RNA sequencing, the precise quantification of gene expression levels and the detection of infrequent variants play a pivotal role in illuminating the intricacies of biological phenomena and disease mechanisms. Nevertheless, several technical obstacles, including PCR duplicates and sequencing errors, impede the fidelity and exactness of RNA-seq data. The advent of Unique Molecular Identifiers (UMIs) has emerged as an influential remedy to overcome these impediments and augment the dependability of analyses conducted in the realm of high-throughput RNA sequencing.
What are UMIs?
Unique Molecular Identifiers (UMIs), also known as molecular barcodes or random barcodes, are short DNA sequences that are incorporated into individual RNA molecules during library preparation for sequencing. Each UMI is designed to be unique and acts as a distinct identifier for its corresponding RNA molecule.
The primary purpose of UMIs in RNA-seq is to enable the accurate identification and removal of PCR duplicates, which are identical RNA fragments that arise due to the amplification process during library preparation. By assigning a unique UMI to each RNA molecule before amplification, researchers can distinguish between genuine biological molecules and PCR duplicates, improving the accuracy and reliability of downstream analyses.
Please refer to our article Digital RNA Sequencing: Introduction, Workflow, and Applications for more details.

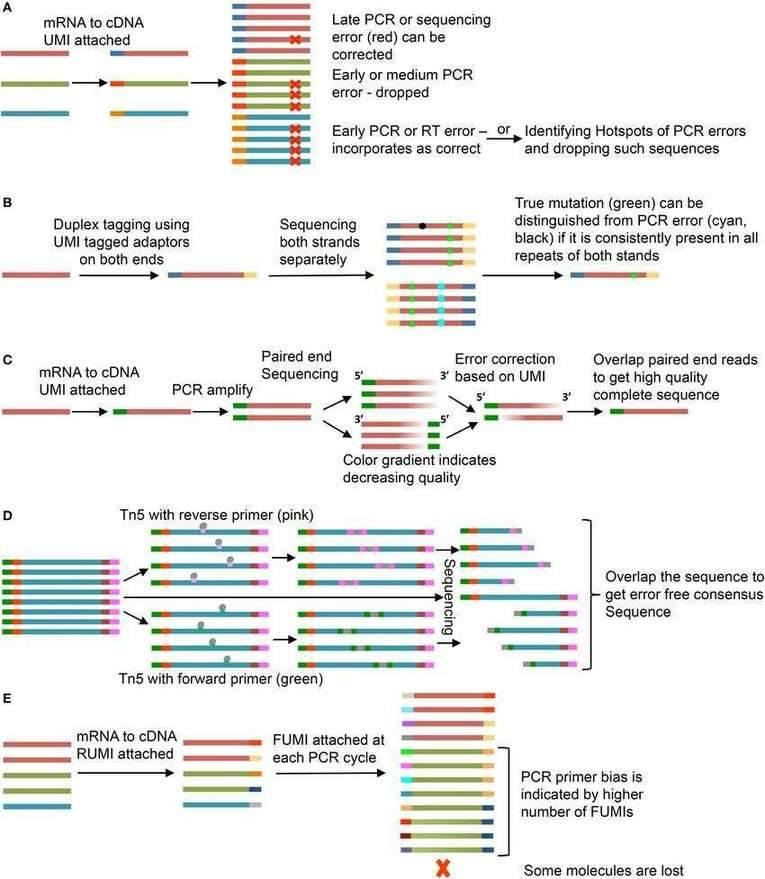
Use of unique molecular identifiers (UMIs). (Chaudhary et al., 2018)
What Are the Differences Between Barcodes and UMIs?
Barcodes and Unique Molecular Identifiers (UMIs) are both used in high-throughput sequencing to label and track individual molecules. While they share similarities in their purpose, there are notable differences between the two.
| Barcodes | Unique Molecular Identifiers (UMIs) | |
|---|---|---|
| Purpose | Sample multiplexing and identification | Address PCR duplicates, enhance quantification accuracy |
| Design | Short sequences (4-12 nucleotides) | Longer sequences (6-12 nucleotides) |
| Application | Enable pooling and identification of multiple samples | Remove PCR duplicates, improve quantification, enhance variant detection |
| Incorporation | Attached to sequencing adapters during library prep | Ligated or attached to RNA or DNA molecules during library prep |
| Usage | Sample indexing for pooled sequencing | Quantification accuracy, variant detection, low-input samples |
| Benefits | Cost and time savings, simultaneous processing of multiple samples | Improved data quality, removal of PCR duplicates, accurate quantification |
| Common Experiments | Multiplexed RNA-seq, multiplexed DNA sequencing for genotyping | RNA-seq, variant detection, low-input samples |
Why We Use UMIs?
Removal of PCR Duplicates
PCR amplification is a crucial step in RNA-seq library preparation, but it can introduce biases and lead to the overrepresentation of certain RNA molecules. PCR duplicates can skew the estimation of gene expression levels and obscure the detection of rare variants. UMIs enable the identification and removal of PCR duplicates by comparing the UMI sequences associated with each RNA fragment. Duplicate reads with the same UMI can be confidently flagged and eliminated, ensuring accurate quantification and reducing bias in downstream analysis.
Accurate Quantification of Gene Expression
UMIs provide a unique identifier for each RNA molecule, enabling precise quantification of gene expression levels. By counting the distinct UMIs associated with a particular gene, researchers can accurately determine the abundance of individual RNA molecules and quantify gene expression with improved accuracy and precision. This is especially valuable for low-input samples or when quantifying low-abundance transcripts.
Detection of Rare Variants
Incorporating gene expression analysis, the utilization of Unique Molecular Identifiers (UMIs) plays a pivotal role in discerning elusive variants within RNA-seq data. Through harnessing the distinct UMI sequences, researchers gain the ability to effectively discriminate genuine biological variations from the inherent errors encountered during sequencing. By facilitating the assembly of consensus sequences from read families sharing identical UMIs, UMIs offer an enhanced capacity to detect variations with heightened sensitivity, particularly when dealing with infrequent mutations or uncommon genetic variants.
Quality Control and Library Complexity Assessment
UMIs also serve as valuable quality control metrics for RNA-seq libraries. By analyzing the distribution of UMIs, researchers can assess library complexity, identifying potential biases or issues that may impact downstream analyses. Evaluating UMI saturation and diversity provides insights into the quality and completeness of the library, allowing researchers to make informed decisions about the reliability of their data.
Challenges of Applying UMIs
Increased Complexity and Cost
Incorporating UMIs into library preparation adds an extra step, which can increase the complexity and cost of the workflow. Additional reagents and protocols are required to introduce and process UMIs effectively.
Potential UMI Misassignment
Despite efforts to design unique UMIs, errors can occur during UMI synthesis or sequencing, leading to UMI misassignment. Misassigned UMIs may result in inaccurate quantification and false identification of PCR duplicates. Careful experimental design, error correction algorithms, and quality control measures are necessary to minimize UMI misassignment.
Computational Challenges
Analyzing data with UMIs requires specific bioinformatics tools and pipelines to process and interpret the sequencing data accurately. Managing and analyzing UMI-tagged reads and implementing appropriate deduplication strategies can be computationally intensive.
Impact on Library Complexity
UMIs themselves can introduce biases if not properly designed or implemented. The UMI sequence length, composition, and method of incorporation may affect library complexity and introduce UMI-specific biases that need to be considered and addressed during analysis.
How To Use UMIs in RNA-Seq Library Prep?
Incorporating Unique Molecular Identifiers (UMIs) into RNA-seq library preparation involves specific steps to ensure the accurate identification and removal of PCR duplicates. Here is a general overview of how to use UMIs in RNA-seq library prep:
- Designing UMI sequences: Determine the length and composition of the UMI sequences based on the specific requirements of your study. UMIs are typically short (6-12 nucleotides) and designed to be unique.
- UMI addition during reverse transcription: Begin the library preparation by adding the UMI sequence to the first-strand cDNA synthesis step. This can be achieved by using a modified reverse transcription primer containing the UMI sequence. The UMI should be ligated or attached to the 5' end of the RNA molecule during this step.
- cDNA synthesis and library preparation: Proceed with reverse transcription, generating cDNA from the RNA template. This cDNA will contain the UMI sequence incorporated at its 5' end. Carry out the library preparation steps, such as fragmentation, end-repair, and adapter ligation, as per the standard RNA-seq library preparation protocol. Ensure that the UMI sequence is preserved throughout these steps.

RNA-Seq Library Prep. (Parekh et al., 2016)
- PCR amplification and UMI retention: Perform PCR amplification to enrich the library and add the necessary sequencing adapters. It is essential to optimize the PCR conditions to minimize PCR biases. During PCR, use primers that anneal upstream and downstream of the UMI sequence to retain the UMI in the amplified products. This ensures that each PCR duplicate will have the same UMI sequence.
- Sequencing and UMI extraction: Sequence the RNA-seq library using a high-throughput sequencing platform. After sequencing, extract the UMI sequence from each read by identifying the position where the UMI is located in the read.
- UMI processing and analysis: Once the UMIs are extracted from the sequencing reads, you can process and analyze the data. The common steps include UMI deduplication, where reads with the same UMI are identified and duplicates are removed, retaining only one representative read. The deduplicated reads are then used for downstream analysis, such as gene expression quantification or variant detection.
References:
- Chaudhary, Neha, and Duane R. Wesemann. "Analyzing immunoglobulin repertoires." Frontiers in immunology 9 (2018): 462.
- Parekh, Swati, et al. "The impact of amplification on differential expression analyses by RNA-seq." Scientific reports 6.1 (2016): 25533.